There are tools such as DSSP https://en.wikipedia.org/wiki/DSSP_(hydrogen_bond_estimation... which will take out the 3d structure determined by crystallography and spit out hte ribbons and helices- for example, for helices, you can see a specific arrangement of carbons along the protein's backbone in 3d space (each carbon interacts with a carbon 4 amino acids down the chain).
Protein motion at room temperature varies depending on the protein- some proteins are rocks that stay pretty much in the same single conformation forever once they fold, while others do thrash around wildly and others undergo complex, whole-structure rearrangements that almost seem magical if you try to think about them using normal physics/mechanical rules.
Having a magical machine that could output the full manifold of a protein during the folding process at subatomic resolution would be really nice! but there would be a lot of data to process.
Certainly, in a corpo like mine (Genentech/Roche) protein structures have a long history of being used in drug discovery- not typically a simple "dock a ligand to a protein" but more for constructing lab experiments that help elucidate the actual mechanistic biology going on. That is only a tiny part of a much larger process to work on disease targets to come up with effective treatments. Genentech is different from most pharma in that their treatments are themselves typically proteins, rather than small molecules.
It seems like that's the basic principle to understand.
In practice, the force fields are well understood but to be computationally efficient, they have to approximate just about everything. Examples: since number of inter-atom distance pairs goes up with N**2 atoms, you need to have tricks to avoid that and instead scale around n log n or even n if you can do it. When I started, we just neglected atoms more than 9 angstrom apart, but for highly charged molecules like DNA, that leads to errors in the predicted structure. Next, typically the force fields avoid simulating polarizability (the ability of an atom's electron cloud to be drawn towards another atom with opposite charge), also because expensive. They use simplified spring models (lterally hooke's equation) for bond lengths, bond angles. The torsions (the angle formed by 4 atoms in a row) haev a simplified form. The interatomic relationships are not handled in a principled way, instead treating atoms as mushy spheres....
After having made major contributions in this area, I don't think that improvements to force fields are going to be the most effective investment in time and energy. There are other bits of data that can get us to accurate structures with less work.
It gives a picture of the enzyme as a machine, and lets you look at specific parts and say “this residue is probably doing this job in the whole system”.
The atoms do wiggle around a bit at room temperature (and even more at body temperature), which means that simulating them usefully typically requires sampling from a probability distribution defined by the protein structure and some prior knowledge about how atoms move (often a potential energy surface fitted to match quantum mechanics).
There are many applications of these simulations. One of the most important is drug design: knowing the structure of the protein, you can zoom in on a binding pocket and design a set of drug molecules which might disable it. Within the computer simulation, you can mutate a known molecule into each of your test molecules and measure the change in binding affinity, which tells you pretty accurately which ones will work. Each of these simulations requires tens of millions of samples from the atomic probability distribution, which typically takes a few hours on a GPU given a good molecular dynamics program.
Some proteins have 3D structures that look like abstract art only because we don't have an intuitive understanding of what shape and amino acids are necessary to convert chemical A to chemical B, which is the main purpose of many enzymes in the body. If you look at structural proteins or motor proteins, on the other hand, their function is clear from their shape.
There are a lot of other things you can do with the shape. If it has a pore, you can estimate the size and type of small molecule that could travel through it. You can estimate whether a binding site is accessible to the environment around it. You can determine if it forms a multimer or exists as a single unit. You can see if protein A and protein B have drastically different shapes given similar sequences, which might have implications for its druggability or understanding its function.
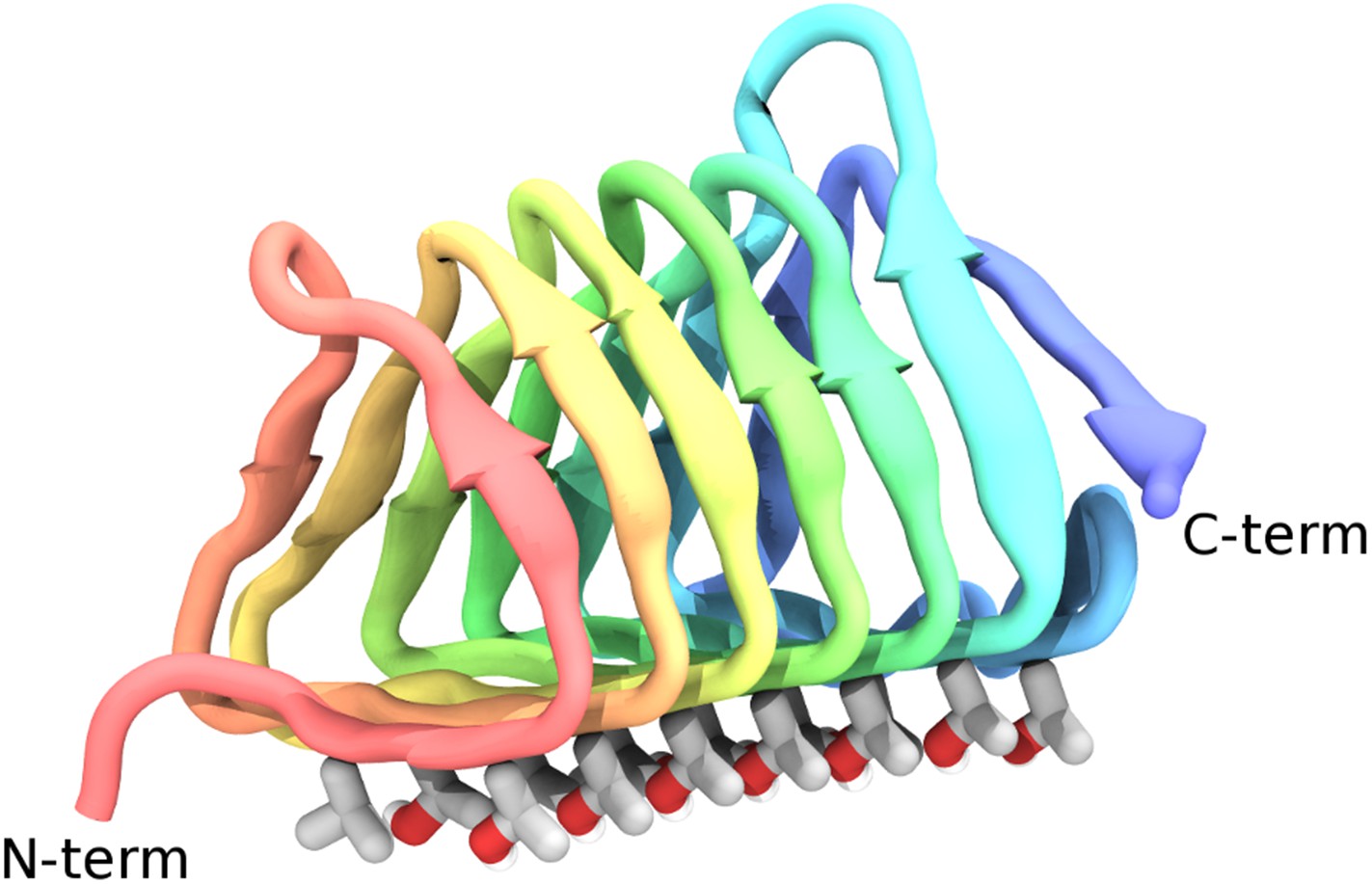
The ribbon shape for GFP is a very cool barrel thing
* https://www.rcsb.org/structure/1m8n
* https://iiif.elifesciences.org/lax/05142%2Felife-05142-fig1-...
{kind=link}
That's one approach.
The thing to understand is that proteins form "binding sites": areas that are more likely to attract other particular regions of proteins or other molecules, or even atoms. Think about hemoglobin. The reason it holds onto oxygen atoms is because it has binding sites.
Binding sites are great because they represent more freedom to do things than molecules typically have. Normal chemistry consists of forming strong electronic bonds between atoms, or forming rigid lattices/crystals.
Binding sites allow molecules to do things like temporarily attach to each other and let each other go under certain circumstances, for instance when another binding site is active/inactive. This can happen through "conformation change", where a molecule bound/unbound on some binding site makes the protein change shape slightly. This is how proteins can act like machines.
> What do the "ribbons" signify
Different regions of the protein have different sequences of amino acids. Amino Acids have somewhat different shapes from each other. The ribbons are actually broader than the spindles (or threads), and less flexible. Not sure about the different colors, maybe someone else can fill in.
> Also, is that what proteins would really look like if you could see at sub-optical wavelength resolutions?
Not really, it's an abstraction. They're big molecules, so if you look closely they're made of atoms, which are (kinda, sorta not really, quantum stuff) spherical.
> So wouldn't they be "thrashing around" like rag doll in blender at room temperature?
Yes, but the attractions between the different parts of the molecule keeps it somewhat under control. So more like an undulating little creature, jellyfish perhaps.
> It seems strange to me that something like that could be so central to life
Yep, gotta remember that it's all statistical. These things are getting made, do their job, breaking, and getting degraded some insane number of times per second. Swarm behavior, sort of.
They certainly do "thrash around", but that thrashing is constrained by the bonds that are formed, which greatly limits the degrees of freedom. Here's a short video of a simulation to demonstrate:
[0]https://learning.edx.org/course/course-v1:MITx+7.00x+2T2022/...
Back in the day, I had steered away from chemistry in college because I didn't like to memorize stuff. Now I realize I missed out on some amazing knowledge.
It's funny you say that, because the first image on the English Wikipedia page for Equipartition Theorem[1] is an animation of the thermal motion of a peptide.
Just like having a human genome sequence, this is not a magic key that solves all problems of biology but a building block for use by researchers. An investigator may look up the folded structure of a protein and use that information to glean certain context-specific insights from it such as how exactly two interacting proteins interact mechanically.
The other significant benefit is that this frees up resources that were spent having to figure out the structure in other ways. It's an efficiency improvement.
This is hugely important for developing drugs and vaccines.
To see the effect of this look no further than prions. Prions are the exact same protein that are folded in weird ways. Worse, they can "transmit" this misfolded shape to other otherwise normal proteins. Prions behave differently just because of the different shape and can lead to disease. This is exactly what Mad Cow's Disease (BSE) is.
What we get taught in high school about chemistry is incredibly oversimplified.
One example of this I like is the geometry of a water molecule. When we first learn about atoms, we learn the "solar system" model (aka Bohr). The reality is instead that we have 3D probability distributions of where electrons might be. These clouds are in pairs. I believe this is to do with the inverted wavefunction really we're getting beyond my knowledge of quantum mechanics here so that's just a guess.
Well those clouds additionally form valence shells. We learn about these and how atoms want to form completely valence shells. So Oxygen has 8 electrons ie 4 pairs of electrons. When bonding with 2 hydrogen atoms we end up with a weird geometry of ~104.5 degrees between the two hydrogen atoms because of how these pairs interact. The naive assumption might expect that the two hydrogen atoms are 180 degree apart.
So back to proteins, you may have learned about hydrogen bonds. This affects molecular shape because when a hydrogen atom shares an electron, it is often positively charged. That positive charge pushes away other positive charges. This is the realy difficulty in protein folding because with a molecule of thousands of atoms and weird geometry you may find distant parts of the molecule interacting with hydrogen bonds.
So a single cell consists of thousands (IIRC) of different proteins. Figuring out those interactions is important but incredibly difficult.
[0] https://en.m.wikipedia.org/wiki/Protein_secondary_structure
Basically, you see these 3D representations of specific proteins as a crumple of ribbons-- literally like someone ran multi-colored ribbons though scissors to make curls and dumped it on the floor (like a grade school craft project).
So... I understand that proteins are huge organic molecules composed of thousands of atoms, right? Their special capabilities arise from their structure/shape. So basically the molecule contorts itself to a low energy state which could be very complex but which enables it to "bind?" to other molecules expressly because of this special shape and do the special things that proteins do-- that form the basis of living things. Hence the efforts, like Alphafold, to compute what these shapes are for any given protein molecule.
But what does one "do" with such 3D shapes?
They seem intractably complex. Are people just browsing these shapes and seeing patterns in them? What do the "ribbons" signify? Are they just some specific arrangement of C,H,O? Why are some ribbons different colors? Why are there also thread-like things instead of all ribbons?
Also, is that what proteins would really look like if you could see at sub-optical wavelength resolutions? Are they really like that? I recall from school the equipartition theorem-- 1/2 KT of kinetic energy for each degree of freedom. These things obviously have many degrees of freedom. So wouldn't they be "thrashing around" like rag doll in a blender at room temperature? It seems strange to me that something like that could be so central to life, but it is.
Just trying to get myself a cartoonish mental model of how these shapes are used! Anyone?